Leaderboard tables compare compounds on an assay-by-assay basis, but they can't tell you which one will work in patients. We built a framework that translates preclinical data into predicted human dose distributions while explicitly propagating uncertainty from experimental measurements and model extrapolation. Applied to three closely matched candidates, it produced a clear ranking that the leaderboard couldn't.
Key takeaways:
- Predicted human dose is the key integrative endpoint for compound prioritization. It combines potency, ADME, and PK into a single, clinically relevant number.
- Dose uncertainty is an intuitive way to quantify risk. Point estimates suggested the three candidates appeared similar; dose distributions showed one was more resilient under pessimistic scenarios.
- Sensitivity analysis identifies the cheapest experiments to reduce risk. In this case, replicate in vitro measurements were a more efficient way to increase projection confidence than more expensive in vivo studies
The problem with leaderboards
For small-molecule drug discovery teams, prioritizing advanced compounds is rarely straightforward. When no single molecule is obviously best along every axis, teams turn to "leaderboard" tables: side-by-side comparisons of potency, ADME, PK, and safety endpoints. Leaderboards highlight differences, but they offer little guidance on which differences actually matter for clinical success. The key question isn't which compound wins the most rows, but how the full set of measurements translates into a likely efficacious dose in humans.
Consider fluconazole and itraconazole, two antifungals with wildly different preclinical profiles (Table 1)1,18. One is small and polar with low clearance, the other large and lipophilic with high clearance and extensive distribution. And yet both achieve long effective half-lives, appreciable bioavailability, and ultimately similar efficacious human doses.
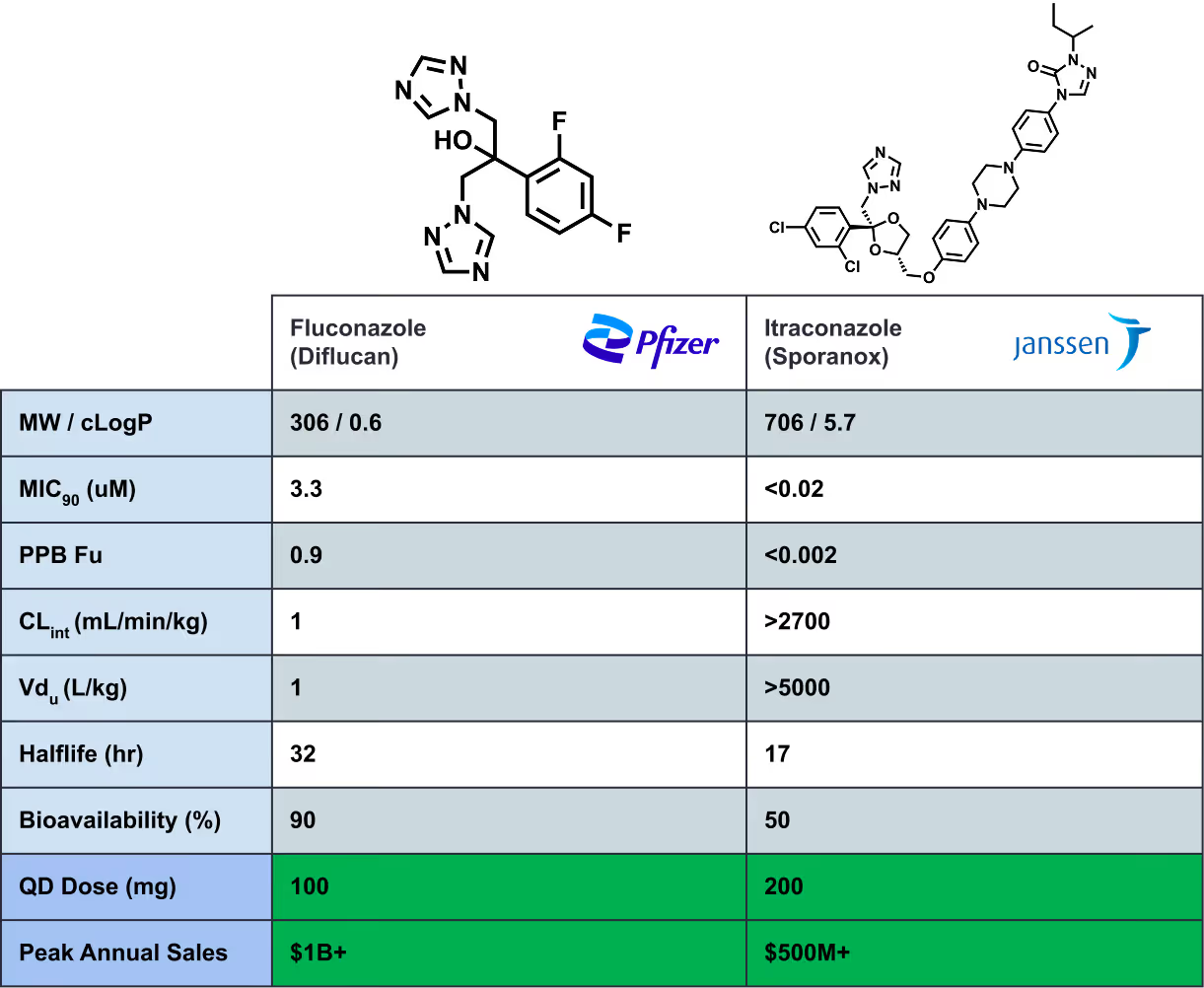
In small-molecule drug discovery, a low efficacious human dose is a key determinant of clinical success1,2. Diflucan and Sporanox both became very successful drugs. Though their profiles look irreconcilable on an assay-by-assay basis, their success emerged from how their properties trade off. The opposite can also be true: compounds with apparently similar leaderboard profiles can diverge meaningfully in predicted human dose. Leaderboards rarely capture these relationships.
That being said, dose projection carries substantial uncertainty due to experimental measurement error, assumptions in the translational model, and inter-patient variability. Point estimates paint an incomplete, often overly optimistic picture. To make rational decisions, teams should compare probabilistic dose projections: quantified ranges of plausible doses that reveal robustness, tail risk, and where additional data would most reduce uncertainty.
This is the approach we took in a recent collaboration with Aleksia Therapeutics. Our framework (1) translates preclinical data into predicted human PK and dose, (2) explicitly propagates both experimental and extrapolation uncertainty, and (3) uses sensitivity analysis to identify the most efficient follow-up experiments to reduce decision risk. The goal is not simply to estimate a single “best guess” dose, but to inform downstream decisions by understanding how uncertainty, bias, and tradeoffs propagate through human PK projections.
Case study: Triaging three advanced candidates at Aleksia Therapeutics
Introduction
Aleksia was evaluating three advanced compounds in the same program during late lead optimization. All three met the program's baseline ADME/PK progression criteria, and each was considered a viable path forward. But the team was operating lean: rather than running exhaustive profiling on every contender, they needed to (1) make the implied human exposure story explicit, and (2) prioritize the most informative follow-up experiments.
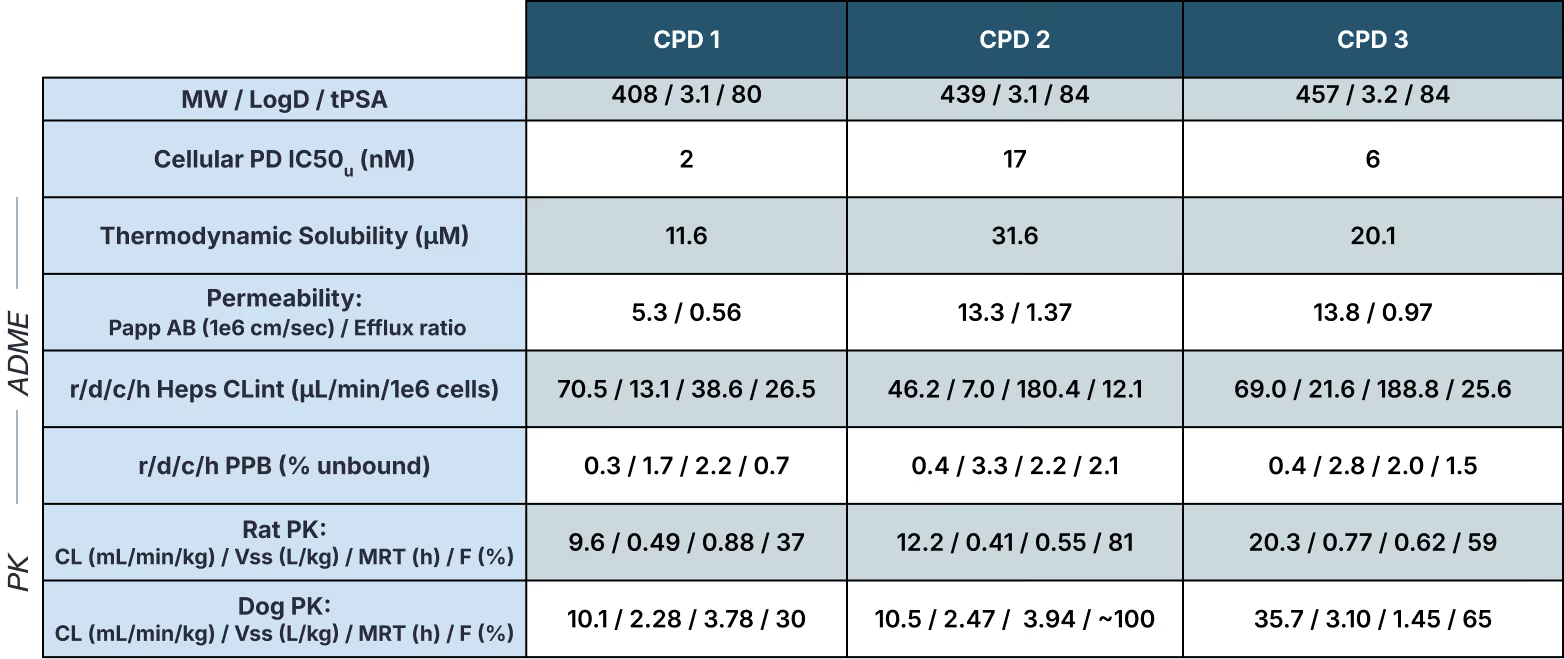
From the leaderboard (Table 2), it wasn't clear which compound would translate to the best predicted human dose. CPD 1 had the strongest cellular potency but the highest human hepatocyte turnover. CPD 2 was less potent but appeared more metabolically stable with higher apparent oral bioavailability. CPD 3 was similar to CPD 1 in potency and stability but had shorter preclinical half-lives.
Each compound had a defensible case depending on which rows you emphasized. But the leaderboard couldn't answer how these tradeoffs would combine in humans, and how robust those conclusions would be given the uncertainty in the underlying measurements and extrapolation techniques. To address this, Aleksia partnered with Inductive to build probabilistic dose projections that explicitly account for uncertainty.
Methodology
(Skip to Results)
We modeled steady-state human PK using a standard 1-compartment model with oral dosing. To keep the comparison focused on the dominant translational uncertainties, we held several assumptions constant across compounds:
- first-order absorption from gut into systemic circulation (ka = 1 h⁻¹)
- blood-to-plasma ratio of 1
- no gut metabolism (Fg = 1)
Available clinical precedent supported a trough-driven efficacy hypothesis, so we targeted an oral twice-daily (BID) regimen maintaining steady-state unbound trough concentration (Cmin,u) above each compound’s unbound IC50 (IC50u).
We estimated clearance using multiple complementary approaches. The primary method was the well-stirred model (WSM): a standard framework that combines hepatocyte intrinsic clearance (CLint), hepatocyte binding8–12, and plasma protein binding to predict in vivo hepatic clearance3–7. To account for possible extra-hepatic-metabolic clearance, we supplemented this with single-species scaling from preclinical CLu13. As a sanity check, an in vitro-to-in-vivo-correlation (IVIVC) analysis on an expanded dataset from the lead series confirmed that in vitro metabolic stability generally predicted in vivo clearance to within ~3x. Where experimental hepatocyte or microsomal binding measurements were unavailable, we used Inductive's Beacon-1 ADMET models to fill the gaps, ensuring consistent estimation across all three compounds.
Human unbound volume (Vss,u) was estimated via unbound volume equivalency from preclinical species14. Bioavailability (F) was predicted both allometrically and by combining the fraction absorbed (Fa), estimated from preclinical equivalency, with predicted hepatic extraction (Fh)15.
How we propagated uncertainty
Single-point estimates of human dose obscure real uncertainty in both the experimental data and the extrapolation methods. We accounted for both sources and propagated them through to predicted human PK (Figure 1).
Experimental uncertainty: We represented variability in each measured input (in vitro potency, ADME, preclinical PK) as distributions informed by biological and technical variability. In the absence of replicates, we used log-normal distributions centered on the measured value with 10th/90th percentiles separated by roughly two-fold.
Extrapolation uncertainty: We incorporated model uncertainty by considering multiple plausible translation paths. For clearance, this meant comparing microsomes vs. hepatocytes, different hepatocyte-binding estimation methods, and multiple sets of physiological scaling factors to convert CLint from in vitro to in vivo units. Each path produced a plausible human CLu distribution which we combined into a final distribution. We constructed analogous distributions for Vss,u, F, and IC50u.
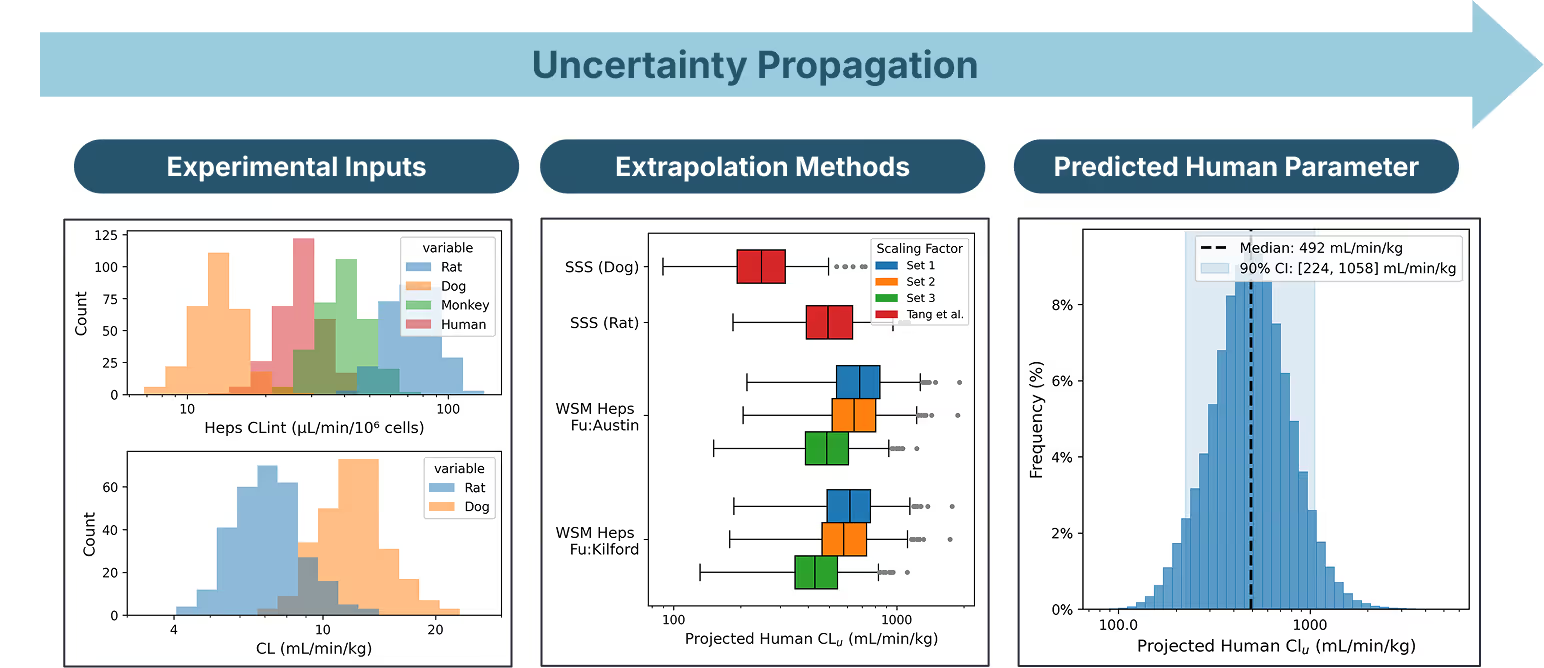
We then used Monte Carlo sampling to draw independent random samples from these parameter distributions. For each draw, we calculated the oral BID dose required to achieve steady-state Cmin,u coverage of the sampled IC50u. This yielded a dose distribution for each compound representing a range of plausible outcomes (Table 3).
Results
CPD 1 was the most robust path forward under uncertainty; not just because it had a lower predicted median dose (200 mg), but because its pessimistic 95th percentile (1.6 g) was substantially lower than CPD 2’s (5 g) and CPD 3’s (12 g). Predicted human half-lives were similar across all three compounds (~3–4 hours), so CPD 1's lower and more tightly bounded dose translated directly into lower predicted Cmax,u and reduced total drug burden.
CPD 2’s median dose (400 mg) remained technically viable given the team’s criteria, but its 90% confidence interval was >70% wider than CPD 1’s. CPD 3 carried the least favorable profile: a median dose of 1.1 g with a 95th percentile of 12 g, making it the highest-risk candidate under pessimistic scenarios.


What drives the uncertainty?
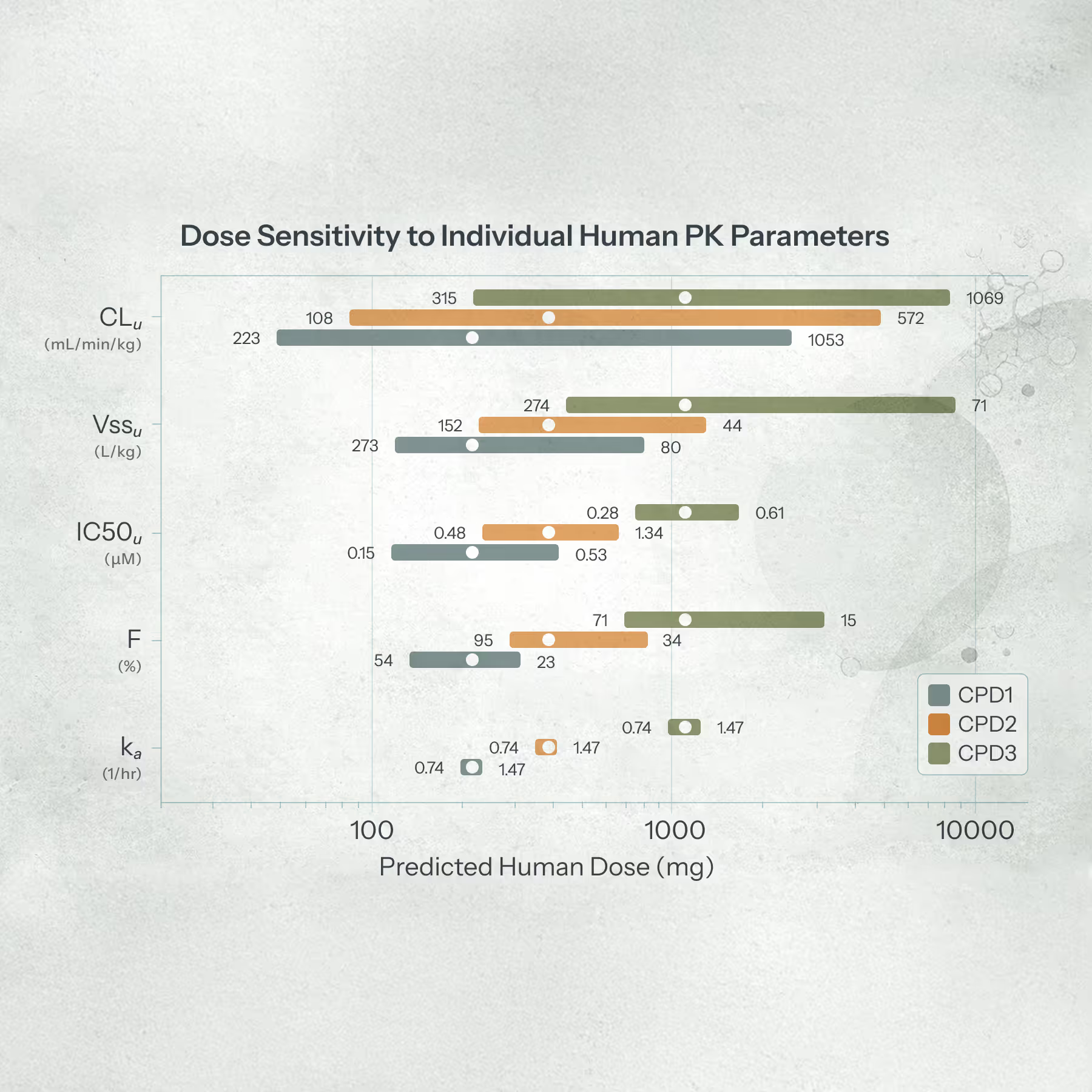
Predicted human clearance was the dominant driver of dose uncertainty across all three compounds. A one-at-a-time sensitivity analysis (varying each PK parameter from its 5th to 95th percentile while holding all others at their medians) showed clearance had the largest impact on predicted dose, followed by volume of distribution, potency, bioavailability, and (with minimal impact) absorption rate (Figure 3).
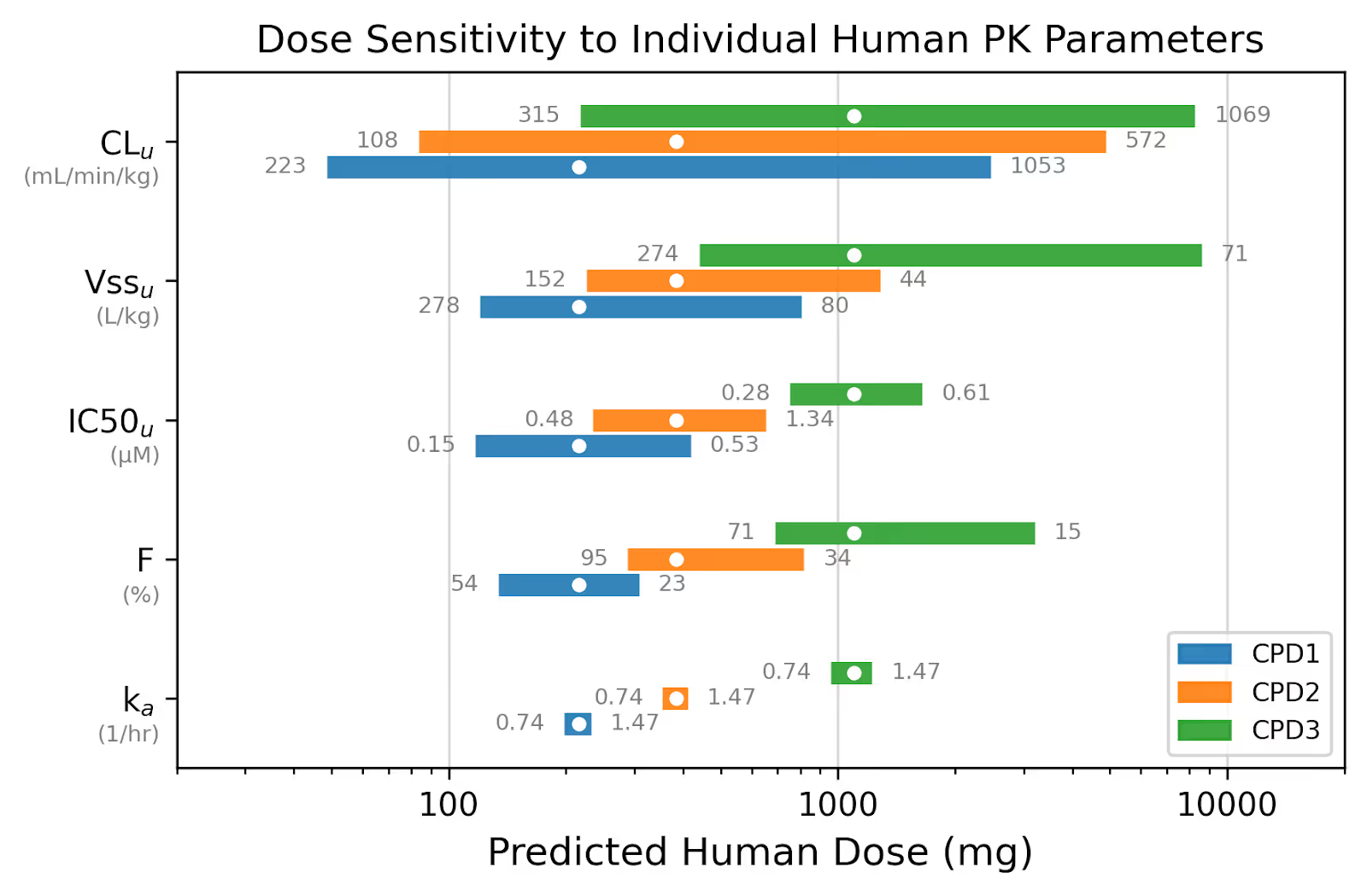
We then drilled into clearance itself, asking: where does the uncertainty in predicted human CL come from? The answer was surprising. Variance in in vitro data (rat and dog plasma protein binding (PPB), and human hepatocyte CLint) contributed as much uncertainty as the more expensive in vivo PK data. Varying human CLint alone shifted the predicted CLu from 370 to 580 mL/min/kg, a >1.5-fold range driven by uncertainty in a single in vitro assay (Figure 4).

Reducing uncertainty cheaply
This finding had a direct experimental implication. With only single measurements for each in vitro assay, we were conservatively modeling wide uncertainty distributions. When we simulated a single “median” replicate for each in vitro assay, tail risk (probability of dose > 1.0 g) was cut in half (from 10% to 5%). The 95% percentile dropped from 1.7 g to 1.1 g (Figure 5), pulling CPD 1 squarely into single-tablet territory with increased likelihood of a simpler formulation and development path than CPDs 2 and 3.
The actionable takeaway: collecting additional replicates of PPB and CLint — cheap, fast in vitro experiments — was the most efficient way to reduce uncertainty in predicted human clearance, and by extension, in predicted human dose. A few weeks of in vitro work costing a fraction of a single PK study could materially derisk the team's compound progression decision.

Assessing predicted safety margins
On the basis of its promising human PK and dose projections, the Aleksia team prioritized CPD 1 for additional off-target profiling. We incorporated the unbound potency for a key off target into our probabilistic dose projection framework to predict the in vivo safety window at efficacious exposures (Figure 6). 95% of simulated outcomes resulted in a Cmax,u safety window ≥ 100x, reassuring the team that they’d have headroom to dose up in the clinic if greater target coverage was needed.

Dose as a decision-ready endpoint
The Aleksia case study illustrates a broader point: the predicted efficacious human dose, when paired with uncertainty, is the closest thing a drug discovery team has to a decision-ready endpoint that integrates key trade-offs into a single framework.
Dose determines total compound burden, shaping on-target coverage and off-target risk. Dose constrains developability: pill burden, dosing frequency, formulation flexibility. All other metrics (potency, ADMET, preclinical PK) are intermediate signals whose clinical relevance is only realized through their impact on dose.
Dose should not be treated as just another multi-parameter optimization (MPO) score. It defines the endpoint that discovery programs are implicitly optimizing toward. Dose-centric thinking can guide decisions across the full arc of a program: qualitatively during lead generation to assess tractability16, quantitatively during lead optimization to arbitrate medicinal chemistry tradeoffs17, and rigorously at development candidate (DC) nomination to manage risk ahead of irreversible investment (Figure 7).

Crucially, this framing does not require pretending that dose predictions are precise or final. The models used here (well-stirred clearance, single-compartment PK, pragmatic scaling) are intentionally simple. They are meant to provide a directionally robust basis for comparing compounds under shared assumptions. More sophisticated approaches are often warranted later, particularly after DC nomination.
But perfection is not the bar for usefulness. Every alternative to dose-centric decision-making (heuristic MPO scores, leaderboard comparisons, or gut feel) also embeds assumptions, biases, and blind spots, usually without making them explicit. Probabilistic dose projection forces teams to confront the question that ultimately matters: how confident are we that we can deliver a safe, efficacious, and developable molecule to patients?
This dose-centric workflow is built into how we work with partners through Embedded Experts, our AI-powered drug discovery consultants. Want to see how it applies to your program? Come talk to us.
Acknowledgements
We thank Hakan Gunaydin, Iain Martin, and Pat Walters for their helpful feedback on this blog post.
References
- Stepan, A. F.; Walker, D. P.; Bauman, J.; Price, D. A.; Baillie, T. A.; Kalgutkar, A. S.; Aleo, M. D. Structural Alert/Reactive Metabolite Concept as Applied in Medicinal Chemistry to Mitigate the Risk of Idiosyncratic Drug Toxicity: A Perspective Based on the Critical Examination of Trends in the Top 200 Drugs Marketed in the United States. Chem. Res. Toxicol. 2011, 24 (9), 1345–1410. https://doi.org/10.1021/tx200168d.
- Nakayama, S.; Atsumi, R.; Takakusa, H.; Kobayashi, Y.; Kurihara, A.; Nagai, Y.; Nakai, D.; Okazaki, O. A Zone Classification System for Risk Assessment of Idiosyncratic Drug Toxicity Using Daily Dose and Covalent Binding. Drug Metab. Dispos. Biol. Fate Chem. 2009, 37 (9), 1970–1977. https://doi.org/10.1124/dmd.109.027797.
- Yang, J.; Jamei, M.; Yeo, K. R.; Rostami-Hodjegan, A.; Tucker, G. T. Misuse of the Well-Stirred Model of Hepatic Drug Clearance. Drug Metab. Dispos. 2007, 35 (3), 501–502. https://doi.org/10.1124/dmd.106.013359.
- Benet, L. Z.; Sodhi, J. K. Can In Vitro–In Vivo Extrapolation Be Successful? Recognizing the Incorrect Clearance Assumptions. Clin. Pharmacol. Ther. 2022, 111 (5), 1022–1035. https://doi.org/10.1002/cpt.2482.
- Tess, D. A.; Ryu, S.; Di, L. In Vitro - in Vivo Extrapolation of Hepatic Clearance in Preclinical Species. Pharm. Res. 2022, 39 (7), 1615–1632. https://doi.org/10.1007/s11095-022-03205-1.
- Hallifax, D.; Foster, J. A.; Houston, J. B. Prediction of Human Metabolic Clearance from In Vitro Systems: Retrospective Analysis and Prospective View. Pharm. Res. 2010, 27 (10), 2150–2161. https://doi.org/10.1007/s11095-010-0218-3.
- Sohlenius-Sternbeck, A.-K.; Jones, C.; Ferguson, D.; Middleton, B. J.; Projean, D.; Floby, E.; Bylund, J.; Afzelius, L. Practical Use of the Regression Offset Approach for the Prediction of in Vivo Intrinsic Clearance from Hepatocytes. Xenobiotica 2012, 42 (9), 841–853. https://doi.org/10.3109/00498254.2012.669080.
- Austin, R. P.; Barton, P.; Mohmed, S.; Riley, R. J. The Binding of Drugs to Hepatocytes and Its Relationship to Physicochemical Properties. Drug Metab. Dispos. 2005, 33 (3), 419–425. https://doi.org/10.1124/dmd.104.002436.
- Austin, R. P.; Barton, P.; Cockroft, S. L.; Wenlock, M. C.; Riley, R. J. The Influence of Nonspecific Microsomal Binding on Apparent Intrinsic Clearance, and Its Prediction from Physicochemical Properties. Drug Metab. Dispos. 2002, 30 (12), 1497–1503. https://doi.org/10.1124/dmd.30.12.1497.
- Kilford, P. J.; Gertz, M.; Houston, J. B.; Galetin, A. Hepatocellular Binding of Drugs: Correction for Unbound Fraction in Hepatocyte Incubations Using Microsomal Binding or Drug Lipophilicity Data. Drug Metab. Dispos. 2008, 36 (7), 1194–1197. https://doi.org/10.1124/dmd.108.020834.
- Gardner, I.; Xu, M.; Han, C.; Wang, Y.; Jiao, X.; Jamei, M.; Khalidi, H.; Kilford, P.; Neuhoff, S.; Southall, R.; Turner, D. B.; Musther, H.; Jones, B.; Taylor, S. Non-Specific Binding of Compounds in in Vitro Metabolism Assays: A Comparison of Microsomal and Hepatocyte Binding in Different Species and an Assessment of the Accuracy of Prediction Models. Xenobiotica 2022, 52 (8), 943–956. https://doi.org/10.1080/00498254.2022.2132426.
- Hallifax, D.; Houston, J. B. Binding of Drugs to Hepatic Microsomes: Comment and Assessment of Current Prediction Methodology with Recommendation for Improvement. Drug Metab. Dispos. 2006, 34 (4), 724–726. https://doi.org/10.1124/dmd.105.007658.
- Tang, H.; Hussain, A.; Leal, M.; Mayersohn, M.; Fluhler, E. Interspecies Prediction of Human Drug Clearance Based on Scaling Data from One or Two Animal Species. Drug Metab. Dispos. 2007, 35 (10), 1886–1893. https://doi.org/10.1124/dmd.107.016188.
- Berry, L. M.; Li, C.; Zhao, Z. Species Differences in Distribution and Prediction of Human VSs from Preclinical Data. Drug Metab. Dispos. 2011, 39 (11), 2103–2116. https://doi.org/10.1124/dmd.111.040766.
- Lucas, A. J.; Sproston, J. L.; Barton, P.; Riley, R. J. Estimating Human ADME Properties, Pharmacokinetic Parameters and Likely Clinical Dose in Drug Discovery. Expert Opin. Drug Discov. 2019, 14 (12), 1313–1327. https://doi.org/10.1080/17460441.2019.1660642.
- Wang, Y.; Wang, W.; Broccatelli, F.; Kenny, J. R.; Wright, M. R.; Sorenson, J.; Desai, P. Human-Focused Multiparameter Optimization Scores for Rank Ordering Compounds during Early Drug Discovery: Validation of PBPK Models Based on Clinical PK Data. J. Med. Chem. 2025, 68 (16), 17960–17970. https://doi.org/10.1021/acs.jmedchem.5c01707.
- Broccatelli, F.; Veeravalli, V.; Cashion, D.; Baylon, J. L.; Lombardo, F.; Jia, L. Application of Mechanistic Multiparameter Optimization and Large-Scale In Vitro to In Vivo Pharmacokinetics Correlations to Small-Molecule Therapeutic Projects. Mol. Pharm. 2024, 21 (9), 4312–4323. https://doi.org/10.1021/acs.molpharmaceut.4c00256.
- Messer, S. A., Jones, R. N., & Fritsche, T. R. (2006). International surveillance of Candida spp. and Aspergillus spp.: report from the SENTRY Antimicrobial Surveillance Program (2003). Journal of clinical microbiology, 44(5), 1782–1787. https://doi.org/10.1128/JCM.44.5.1782-1787.2006

Quantitation in Combination

Lessons from the Polaris ADMET competition
